


Abstract
Background Although total disc replacement has been performed for years outside the United States, relatively little available data address clinical outcomes, particularly data from prospective studies. We report the 24- to 36-month follow-up of one center's experience with the ProDisc-L artificial disc as part of a prospective, randomized trial comparing total disc arthroplasty to combined anterior–posterior lumbar fusion.
Methods The study involved clinical results for 157 patients from a single center enrolled in the US Food and Drug Administration–regulated trial comparing ProDisc-L to fusion. Only patients who had reached a minimum 24-month follow-up were included in the study. Patients were randomized to receive total disc replacement or circumferential fusion at 1 or 2 lumbar disc levels from L3 to S1, with specific inclusion and exclusion criteria. Data were collected preoperatively and at 6 weeks to 36 months postoperatively. The primary clinical outcome measures were Visual Analog Scale (VAS) scores to assess pain and Oswestry Disability Index (ODI) scores to measure function.
Results The VAS and ODI scores in both treatment groups improved significantly as early as the 6-week followup visit and remained significantly improved throughout the 36-month follow-up period. Although a tendency was observed for the ProDisc-L scores to indicate more favorable outcome, the differences were not statistically significant. The proportion of patients who would have the same procedure again was greater in the total disc replacement group at all follow-up intervals, and significantly greater at the 6- month, 12-month, 24-month, and 36-month follow-up visits.
Conclusions The results of this study indicate that the total disc replacement with ProDisc-L produces improvements in pain and function that are at least as good as those provided by circumferential fusion. During the long-term follow-up of 24 and 36 months, outcomes did not become less favorable compared with the early outcomes.
Clinical Relevance We found that results of total disc replacement were at least as good as those achieved with combined instrumented anterior–posterior fusion for the treatment of painful disc degeneration. Favorable results were maintained during 24- and 36-month follow-up.
INTRODUCTION
The ideal treatment for symptomatic disc degeneration remains elusive. As with many spinal conditions, nonoperative management should be the first line of treatment. If this fails to provide adequate relief, operative intervention may be warranted in carefully selected patients. In a randomized study, the Swedish Lumbar Spine Study Group found that fusion yielded superior results to nonoperative care.1 However, the results of fusion in general remain highly variable, and the conclusions of many studies are difficult to interpret or generalize because of the mix of diagnoses included within a study, the variation of graft materials, instrumentation, combinations of interbody and posterior procedures, and outcome measures used.
In a comprehensive review by Bono and Lee, it was reported that although there was a significant increase in the use of various instrumentation systems for spinal fusion surgery, no correlative improvements in fusion rates or clinical outcome was identified.2 Potential problems with fusion include pseudoarthrosis and adjacent segment deterioration, although the incidence of clinically significant changes at adjacent segments is not well documented. Additionally, there is not a direct correlation between radiographic fusion and functional outcome. Radiographic fusion rates tend to be greater than the rate of favorable clinical outcome. This may suggest that the rate of fusion determined from radiographs is erroneously high or that clinical outcome may be poor even in patients in whom a technical success of bony union was achieved.
Joints should allow motion. Motion-retaining spinal implants are intuitively appealing because they provide the potential for motion at the operated segment. Total disc replacement (TDR) has been used in Europe since the mid-1980s3 and the results from the European experience have been favorable,3–10 even in long-term follow-up of 7 to 10 years.6, 8 However, these European studies were not designed to be prospective trials using randomization to treatment and control groups. The results of a large prospective trial in the United States comparing the Charité artificial disc (DePuy Spine, Raynham, Mass) to anterior lumbar interbody fusion found that TDR was similar to, or better than, fusion.11 Preliminary results with 6- month follow-up in a study comparing ProDisc-L (Synthes Spine, West Chester, Pa) to combined anterior–posterior fusion had similar results.12 The purpose of our study was to analyze results of TDR with ProDisc-L during 24- to 36-month followup in a prospective, randomized trial. This trial compared this treatment to combined anterior–posterior fusion at 1 or 2 disc levels in the treatment of symptomatic disc degeneration unresponsive to nonoperative management.
MATERIALS AND METHODS
This report presents data from the largest-enrolling single center participating in the US Food and Drug Administration (FDA)–regulated investigational device exemption trial evaluating ProDisc-L. At each site, the first 3 patients enrolled received the ProDisc-L. Thereafter, patients were randomized to either ProDisc-L or a combined anterior–posterior instrumented fusion. The randomization was performed according to a 2:1 ratio of ProDisc-L to fusion procedures. Separate randomization tables were used for the single- and double-level procedures. The study was approved by the governing institutional review board and written informed consent was obtained from all patients.
At the time of this report, 157 patients enrolled in the trial had completed 24- to 36-month follow-up. A description of the study population is provided in Table 1. This group included 3 training cases who received ProDisc-L, 89 randomized cases (59 to ProDisc-L, 30 to fusion), and 65 continued-access patients (continued access is a separate phase of the FDA investigational device exemption study that allows ongoing enrollment of patients to receive the investigational device after enrollment in the randomized phase has been completed). The continued-access patients were included in the current study because their selection criteria were identical to those of the randomized study. The data from 1 patient randomized to ProDisc-L were deleted from the analysis. In this patient with 2-level disease, a ProDisc-L was implanted at the L5–S1 level. Upon preparing the L4–L5 level, a large cyst was discovered in the L4 vertebral body. It was opted to do a circumferential fusion at this level. The number of levels operated, as well as the specific levels operated in the 2 surgery groups are presented in Table 1. Thirty-six month follow-up data were available for a subgroup of 48 patients.
Summary of Study Population Characteristics: Total Disc Replacement (TDR) and Fusion Groups
Inclusion and exclusion criteria for this FDA study have been published previously in great detail.12 The primary criteria for study enrollment were degenerative disc disease at 1 or 2 levels, at least 6 months of failed nonoperative treatment, age from 18 to 60 years, Oswestry Disability Index (ODI)13 score of at least 40%, and psychological ability to comply with the study. The primary exclusion criteria were disease at more than 2 levels, prior lumbar spinal fusion, clinically significant facet joint degeneration, degenerative spondylolisthesis greater than Grade I, osteoporosis, and morbid obesity.
Perioperative data collected and compared for this study included operative time, blood loss, and length of hospital stay. Clinical outcome data were collected prior to surgery and 6 weeks, 3 months, 6 months, 12 months, 18 months, 24 months, and 36 months after surgery. Clinical outcome measures were Visual Analog Scale (VAS) to assess pain and postoperative satisfaction with the results of the surgery, modified ODI, and a question asking patients if they would have the same surgery again (response of “yes,” “no,” or “maybe” was to be checked by the patient). Clinical, neurologic, and radiographic parameters were also collected at each study visit.
The surgical techniques for the treatment and fusion control groups have previously been published in detail elsewhere.12 All ProDisc-L procedures used a retroperitoneal approach performed by an access surgeon. In the fusion group, the anterior procedure was performed with the same approach to implant a femoral ring allograft into the disc space. After completion of the anterior procedure, an instrumented intertransverse fusion was performed posteriorly. Autogenous iliac crest bone graft was used in all cases and was generally combined with bone graft–extender material. Unilateral or bilateral instrumentation with pedicle screw fixation was used in all cases.
STATISTICAL METHODS
The TDR and fusion groups were compared with independent sample t test based on the VAS and ODI assessments. Pre- to postoperative scores on the VAS and ODI evaluations were compared with paired t tests. Although repeated-measures analysis of variance may have been preferable, cases would have been excluded if the patient had missed any of the follow-up visits. For the results dealing with categorical response data, such as the satisfaction assessment that asked if patients would have the surgery again with responses of “yes,” “no,” or “maybe,” a χ2 test was used to compare the proportional distribution of responses at each follow-up period in the TDR and fusion groups.
RESULTS
Perioperative Data
As presented in Table 2, TDR was associated with significantly less blood loss, operative time, and length of hospital stay compared to fusion (P < .01 for all comparisons).
Mean Operative Time, Blood Loss, and Length of Hospital Stay in the Total Disc Replacement (TDR) and Fusion Groups
Clinical Outcome
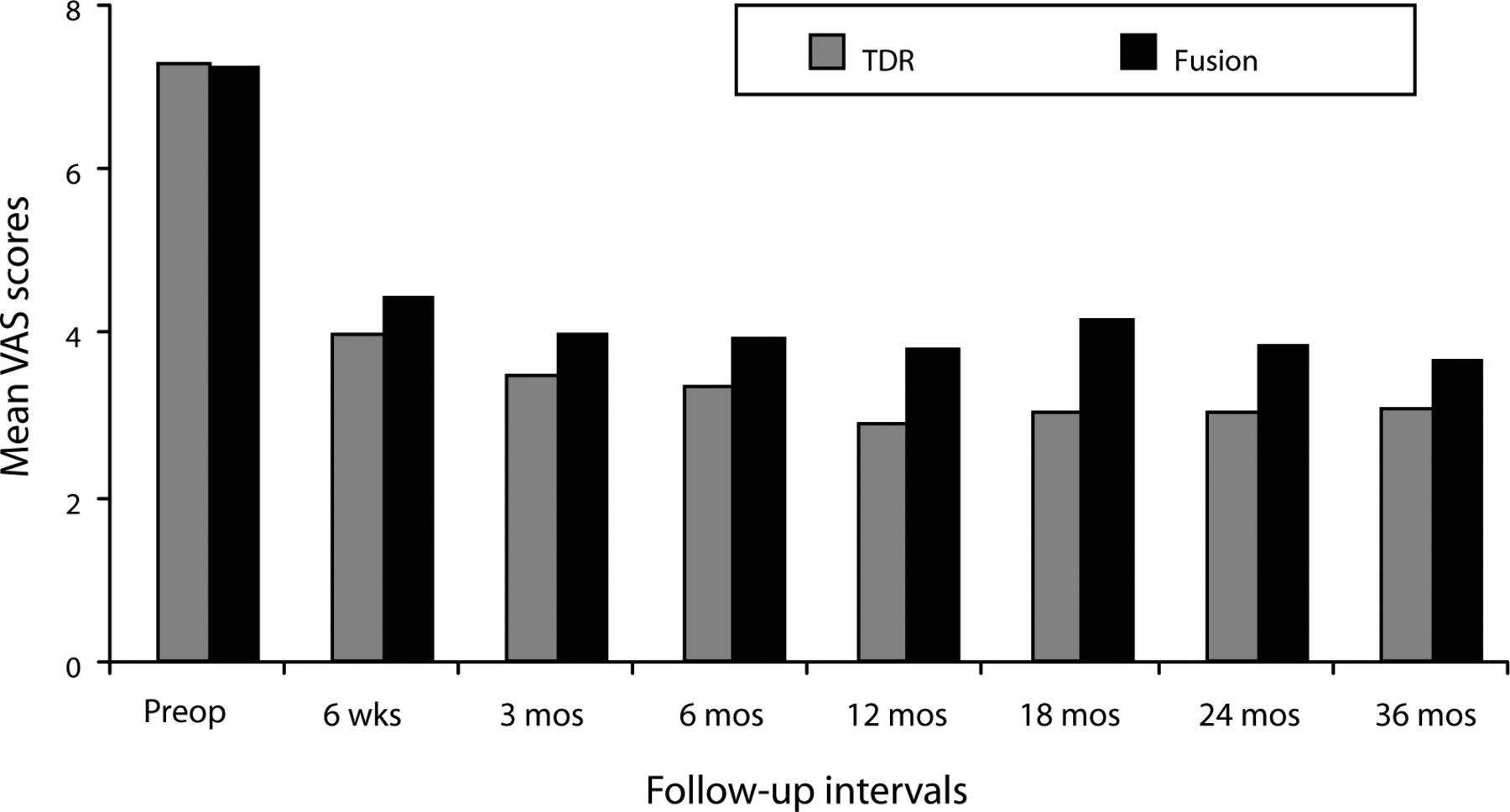
In both the ProDisc-L and fusion groups, mean VAS scores assessing pain improved significantly at the 6-week follow-up from the preoperative value and remained improved at each follow-up interval throughout the 36 months (P < .001; Figure 1). At all follow-up intervals, mean ProDisc-L scores were less than the fusion scores, but the differences were not statistically significant.
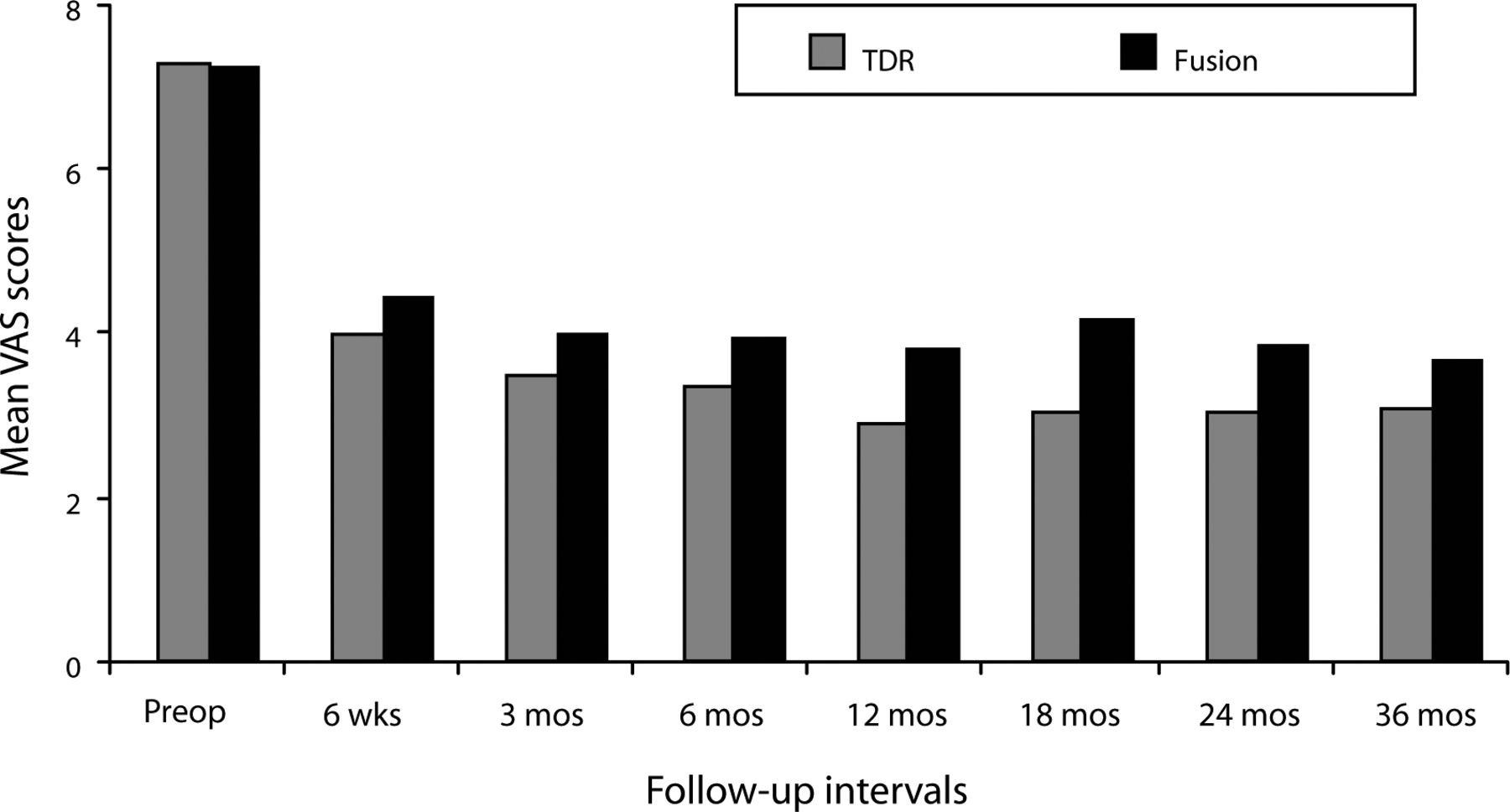
Mean Visual Analog Scale (VAS) scores assessing pain decreased significantly in both surgical groups by the 6-week follow-up visit and remained significantly less than the preoperative value throughout the remainder of the 36-month period. Although mean total disc replacement (TDR) VAS scores were less than the fusion scores at each follow-up period, differences were not statistically significant.
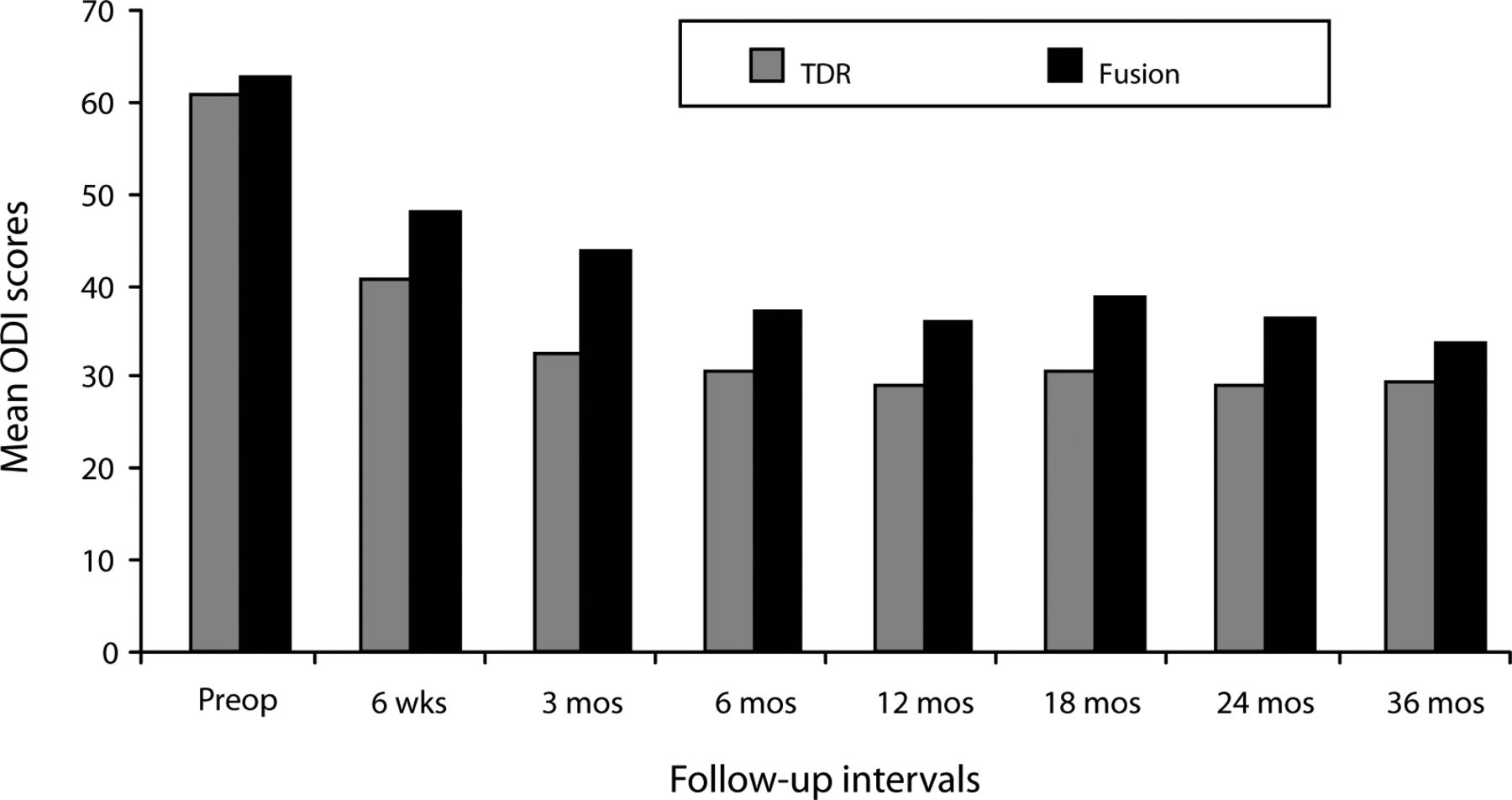
Similar results were noted on the ODI scores. In both surgical groups, mean scores significantly improved by the 6-week follow-up compared with the preoperative mean score, and follow-up values remained significantly improved throughout the 36-month period (P < .05; Figure 2). At all follow-up intervals, the mean ProDisc-L scores were less than the fusion scores, but the difference was statistically significant at only the 3-month follow-up (P < .05).
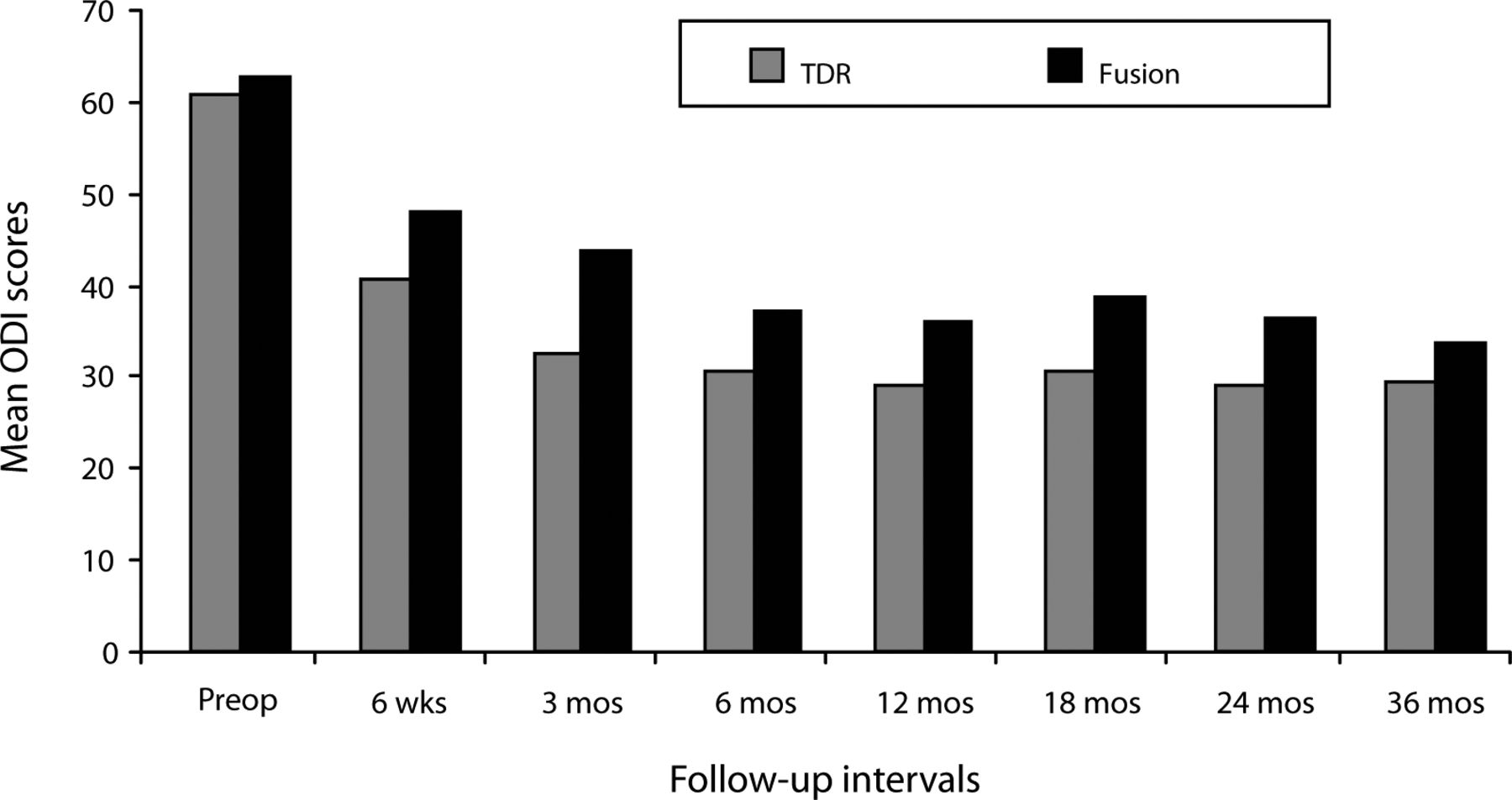
Mean Oswestry Disability Index (ODI) scores decreased significantly in both surgical groups by the 6-week follow-up visit and remained significantly less than the preoperative value throughout the remainder of the 36- month period. Although mean total disc replacement (TDR) ODI scores were less than the fusion scores at each follow-up period, the difference was statistically significant only at the 3-month follow-up.
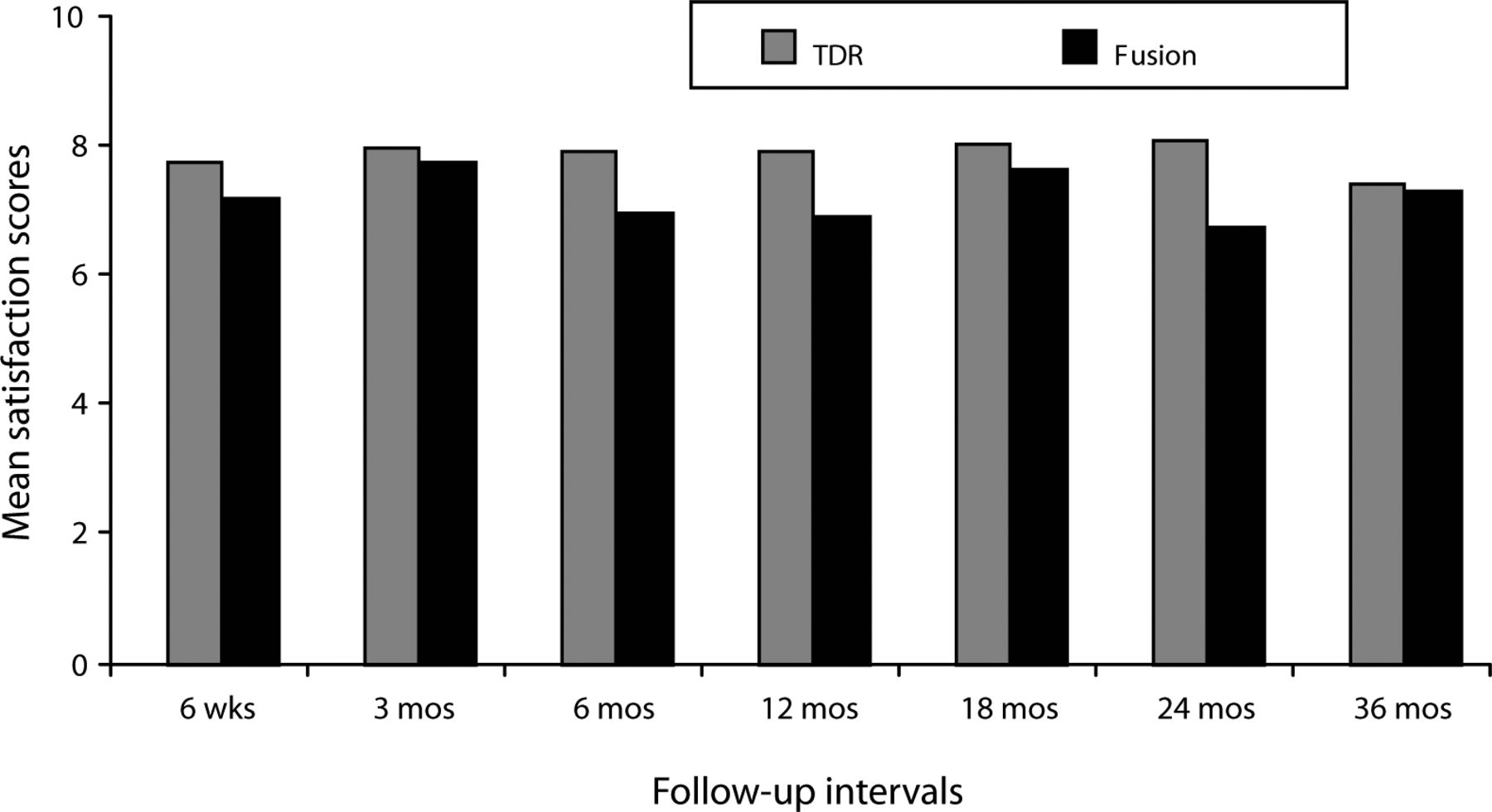
With respect to patient satisfaction, scores for both treatment groups were high (Figure 3), with the TDR group scores being higher at all follow-up periods, although only the difference in 24-month follow-up scores was statistically significant.

Mean postoperative satisfaction scores (greater scores indicate greater satisfaction on a 0 to 10 VAS scale) were high in both groups. Mean scores in the total disc replacement (TDR) group were greater at all visits, but statistically significantly so at the 24-month follow-up (P < .05).
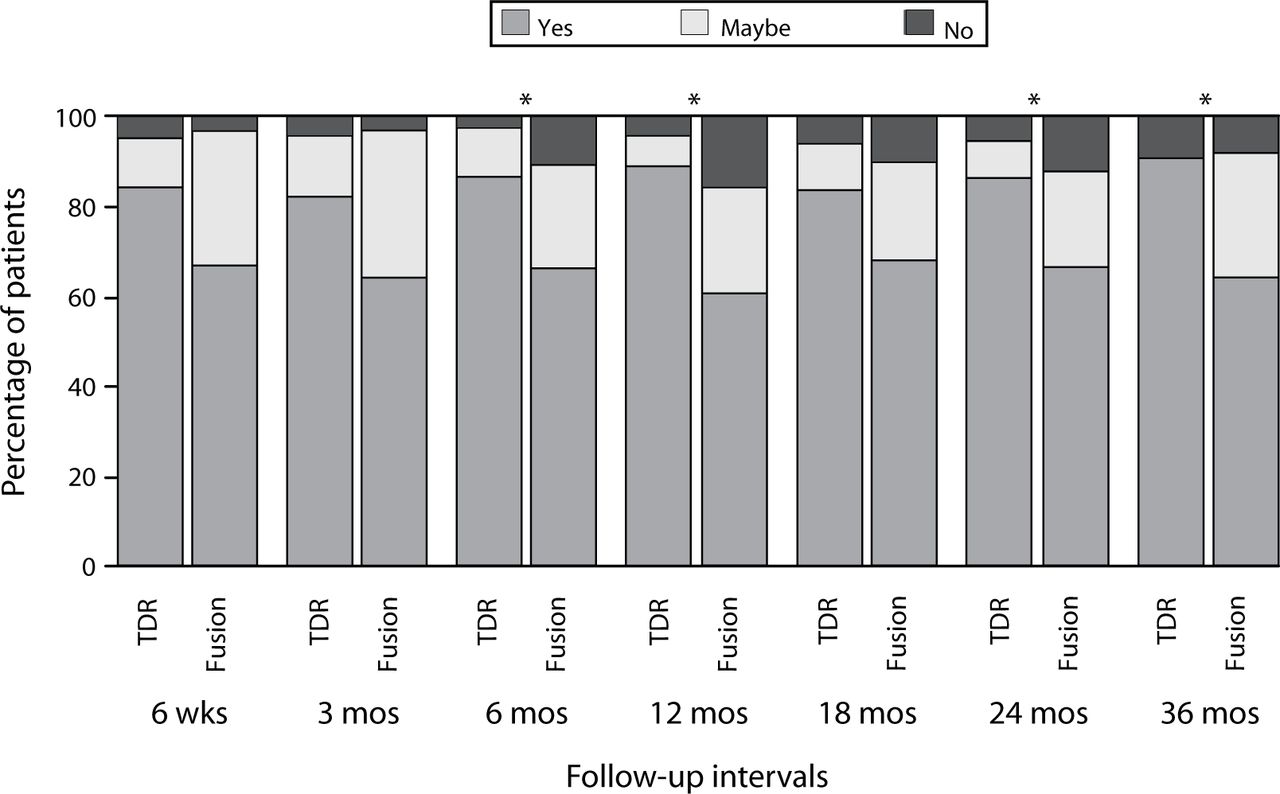
Another measure of satisfaction was assessed: At each follow-up period patients were asked the question, “Remembering the pain you felt before your surgery, would you have this surgery again?” and were instructed to select “yes,” “no,” or “maybe.” At the 6-, 12-, 24-, and 36-month periods, the responses were significantly more favorable in the ProDisc-L group than in the fusion group (Figure 4; P < .05; χ2).

Responses to the question “Remembering the pain you felt before your surgery, would you have this surgery again?” Values presented are the percentage of patients selecting the indicated response at each of the follow-up periods. At the 6-, 12-, 24-, and 36-month periods, the distribution of the responses was significantly more favorable in the ProDisc-L group (P < .05;χ2).
DISCUSSION
This study, based on one center's experience participating in a prospective, randomized, multicenter FDA-regulated noninferiority trial, found that patients in the ProDisc-L group had outcomes at least as favorable as fusion throughout the 24- to 36-month follow-up periods. At no time were the mean scores worse for TDR than for fusion. The proportion of patients responding that they would have the same surgery again was significantly greater among the TDR patients at most of the follow-up periods.
The results of the VAS pain scores and ODI questionnaire in this study were very similar to those in the Charité artificial disc trial reported by Blumenthal et al. in the multicenter investigational device exemption trial for that device, which involved 304 patients with 24-month follow-up.11
Our study demonstrated that significant improvement was noted at the initial 6-week follow-up visit and was consistently maintained throughout the remainder of the 36-month followup. In a recent report by Tropiano et al. that investigated the 7- to 11-year outcome, mean 8.7 years, of TDR with the ProDisc-L device in France, significantly improved back pain, leg pain, and functional scores were found at the long-term follow-up.8 These authors reported that no device-related complications occurred during the study. In a study of minimum 10-year follow-up of the Charité device, Lemaire et al. used a relative gain analysis from pre- to postoperative status to determine long-term outcome.6 They reported 62% of patients with an excellent result and an additional 28% of patients with good outcomes.
Blood loss, operating time, and length of hospital stay were all significantly less in the ProDisc-L group than in the fusion group. This is likely attributable to the inclusion of an instrumented posterior procedure in the fusion group. The fact that similarto- superior results were obtained in the TDR group without requiring a posterior procedure would appear to lend support to the use of these devices.
Our study was based on the 24- to 36-month follow-up of patients enrolled in a prospective, randomized study comparing TDR with ProDisc-L to combined anterior–posterior fusion at 1 or 2 levels in the lumbar spine. The results of this noninferiority study found that TDR produced outcomes no worse than, and in some cases superior to, outcomes for fusion, the traditional treatment for symptomatic disc degeneration unresponsive to nonoperative management. Disc replacement was associated with significantly less blood loss, operative time, and hospital stay. During the long-term follow-up, there was no indication of results deteriorating. These findings suggest that TDR is a viable treatment for symptomatic disc degeneration and maintains acceptable results in multiyear follow-up.
Footnotes
The study was conducted under a protocol approved by Presbyterian Hospital of Dallas, Texas.
- Received December 7, 2006.
- Accepted March 28, 2007.
- Copyright SAS - Spine Arthroplasty Society 2007
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
One or more of the authors or the institutions with which they are affiliated has the following disclosures: consulting agreement, funding for travel to lecture, and research and education support from the manufacturer of the ProDisc (Synthes, West Chester, Pa), and has received funding in excess of $1000.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.